Introduction
An elimination reactions (E1 and E2 reactions) is a type of organic reaction in which two substituents are removed from a molecule in either a one- or two-step mechanism. The one-step mechanism is known as the E2 reaction, and the two-step mechanism is known as the E1 reaction. The numbers refer not to the number of steps in the mechanism, but rather to the kinetics of the reaction. E2 is bimolecular (second-order) while E1 is unimolecular (first-order). In cases where the molecule is able to stabilize an anion but possesses a poor leaving group. In this article we will see detailed mechanism of E1 and E2 reactions, ozonolysis, Markovnikov’s and Anti-Markovnikov Orientation and free radical addition reactions of alkenes.
E1 reactions
The E1 reaction (Unimolecular Elimination) proceeds through a carbocation intermediate. It is also known as the second order reaction. It involves the loss of a leaving group (usually a halide) and subsequent deprotonation. The overall process results in the formation of a double bond. The rate of the E1 reaction depends only on the concentration of the substrate (alkyl halide). Varying the base concentration (e.g., water) doesn’t affect the rate. This suggests that the base is not involved in the rate-determining step. The reaction rate is fastest for tertiary substrates, slower for secondary substrates, and slowest for primary substrates.
Mechanism
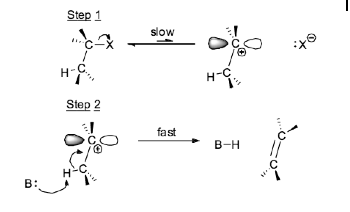
Step 1: Leaving Group Departure
The alkyl halide (substrate) loses the leaving group (e.g., Br) to form a carbocation. This step is usually the rate-determining step.
Step 2: Deprotonation
A weak base (often a solvent molecule) abstracts a proton from an adjacent carbon, creating a double bond. The resulting product is an alkene.
Kinetics
- E1 reactions follow first-order kinetics. This means that the rate depends solely on the concentration of the substrate.
- The rate equation can be expressed as: Rate = k[substrate].
- Deuterium isotope studies provide further evidence for the E1 mechanism.
Comparison with E2
- E1 reactions differ from E2 reactions (Bimolecular Elimination) in that E1 proceeds through a carbocation intermediate, while E2 occurs in a single step.
- E2 reactions require a strong base, whereas E1 reactions only need a weak base.
Order of Reactivity
- Alkyl halides follow the order of reactivity: RI > RBr > RCl.
- Tertiary alkyl halides (those with three alkyl groups attached to the carbon bearing the halogen) are the most reactive in E1 reactions. Because they readily form stable carbocation intermediates during ionization.
E2 reactions
The E2 reaction (Bimolecular Elimination) occurs in a single step. It involves the simultaneous removal of a leaving group (usually a halide) and the abstraction of a proton by a base. The result is the formation of a double bond (alkene).
Key Points
- Concerted Mechanism: E2 reactions proceed via a concerted mechanism, meaning that the bond-breaking and bond-forming steps happen simultaneously. The base (OH⁻) swoops in, abstracting a proton (H) from the carbon adjacent to the leaving group. As the proton leaves, the base attacks the nearby carbon, aiding in the removal of the leaving group. A double bond (alkene) forms between the two carbon atoms.
- Transition State: The transition state involves the anti-periplanar arrangement of the hydrogen (H) and the leaving group (X). This arrangement ensures efficient overlap of orbitals during bond formation. The transition state resembles a molecular ballet. The C-H and C-Br bonds are partially broken, while the O-H and C=C π bond are partially formed. The negative charge is shared between the base and the leaving group, each bearing a partial negative charge.
Stereochemistry
- E2 reactions are stereospecific. They favor the anti-periplanar arrangement of the hydrogen (H) and the leaving group (X).
- This arrangement ensures efficient orbital overlap during bond formation.
Kinetics
- E2 reactions follow second-order kinetics.
- The rate depends on the concentrations of both the alkyl halide (substrate) and the base.
- Mathematically, the rate equation can be expressed as: Rate = k[substrate][base].
- Deuterium isotope studies provide evidence for the E2 mechanism:
- Deuterated substrates (where C–H bonds are replaced by C–D bonds) react at different rates compared to non-deuterated substrates.
- The higher energy required to break the C–D bond (compared to C–H) affects the overall reaction rate.
Order of Reactivity
In E2 reactions, reactivity increases with the number of alkyl groups on the substrate:
- Tertiary alkyl halides are the most reactive.
- Secondary alkyl halides come next.
- Primary alkyl halides are less reactive.
Comparison of E1 and E2 reactions
| Feature | E1 Reaction | E2 Reaction |
| Mechanism | Two-step process: 1. Formation of carbocation 2. Deprotonation to form double bond | Single-step process: Base abstracts a proton as leaving group departs |
| Kinetics | First-order: Rate depends only on concentration of substrate | Second-order: Rate depends on concentration of both substrate and base |
| Substrate | Prefers tertiary alkyl halides due to stable carbocation formation | Prefers tertiary alkyl halides, but primary can also react if the base is sterically hindered |
| Base | Weak base (often the solvent) | Strong base required |
| Solvent | Polar protic solvents which stabilize carbocations | Polar aprotic solvents which do not stabilize carbocations |
| Leaving Group | Departs before the rate-determining step, forming a carbocation | Departs simultaneously with proton abstraction |
| Regioselectivity | Zaitsev’s rule often observed (formation of most substituted alkene) | Zaitsev’s rule often observed, but can be overridden by steric or electronic factors |
| Stereochemistry | Not stereospecific due to planar carbocation intermediate | Stereospecific; requires anti-periplanar geometry between H and leaving group |
Factors affecting E1 and E2 reactions
Elimination reactions are captivating processes where two substituents are removed from a molecule, resulting in the formation of a double bond. Here’s what you need to know:
Base Strength
- E2 Reaction: Strong bases favor the E2 mechanism. These bases are typically alkoxides (such as hydroxide or ethoxide) or amides (like sodium amide). The strong base abstracts a proton (H) from the substrate, leading to the formation of the double bond.
- E1 Reaction: E1 mechanisms only require a weak base. The rate-limiting step in E1 reactions is the formation of a carbocation intermediate. Since the base doesn’t directly participate in the rate-determining step, its strength is less critical.
Solvent
- E1 Reaction: Good ionizing solvents (polar protic solvents) favor the E1 mechanism. Examples include water (H₂O), alcohols (ROH), and carboxylic acids. These solvents stabilize the carbocation intermediate by solvating it.
- E2 Reaction: E2 reactions are favored by polar aprotic solvents. These solvents (such as acetone or dimethyl sulfoxide) do not have acidic protons and do not solvate the base. They allow for efficient base attack on the substrate.
Substrate (Alkyl Halide)
Primary Alkyl Halides
These compounds can undergo both E1 and E2 reactions. Distinguishing between the two pathways is essential.
Secondary and Tertiary Alkyl Halides
- E2 Reaction: These substrates predominantly follow the E2 pathway due to the availability of a strong base.
- E1 Reaction: Tertiary alkyl halides favor E1 reactions because the stability of the carbocation intermediate plays a crucial role. More stable carbocations (tertiary > secondary > primary) lead to faster E1 reactions.
Stereochemistry
- E2 Reaction: The stereochemistry matters significantly. The hydrogen being removed must be anti to the leaving group (i.e., they must be in a trans arrangement). This antiperiplanar alignment allows efficient base attack.
- E1 Reaction: E1 reactions tolerate various stereochemistries. The hydrogen can be oriented in any direction relative to the leaving group.
Heat
Both E1 and E2 reactions are favoured by elevated temperatures. Heat provides the necessary activation energy for these elimination processes.
Carbocation Rearrangements
A carbocation is a positively charged carbon species (C⁺) with only three bonds and an empty p orbital. It’s a reactive intermediate that forms during various organic reactions. Carbocations are often unstable due to their positive charge, but they can rearrange to more stable forms.
Why Do Carbocations Rearrange?
Carbocations rearrange because of stability issue. When a carbocation shifts its structure, it aims to minimize charge separation and achieve a more favourable arrangement of substituents. The driving force behind rearrangement is the quest for greater stability.
Types of Carbocation Rearrangements
Hydride Shift
- In a hydride shift, a hydrogen atom (H⁻) migrates from an adjacent carbon to the positively charged carbon center.
- This shift results in a more stable carbocation.
- Hydride shifts often occur in various types of carbocations.
Alkyl Shift
- An alkyl group (such as a methyl or ethyl group) moves from one carbon to another within the carbocation.
- Alkyl shifts also contribute to stabilizing carbocations.
- These rearrangements are common in organic reactions.
Evidence for Carbocation Rearrangements
Product Formation
- When a carbocation rearranges, it leads to a different product than the initially formed carbocation.
- The major product observed often corresponds to the rearranged carbocation.
Saytzeff’s Rule (Zaitsev’s Rule)
- Named after the Russian chemist Aleksandr Zaitsev, this rule states that during elimination reactions (such as dehydrohalogenation), the more substituted alkene (Zaitsev product) is favored.
- Specifically, a hydrogen is removed from the β-carbon (the carbon adjacent to the leaving group) with the fewest hydrogens.
- This preference arises from the greater stability of the more substituted alkene due to hyperconjugation effects.
- Saytzeff’s orientation aligns with the idea that stability drives product formation.

Stability Factors for Carbocations
- Adjacent Electron-Withdrawing Groups: Carbocations are stabilized when adjacent to elements with lone pairs (e.g., oxygen, nitrogen, sulfur). These elements share their electron pairs, leading to resonance stabilization. Allylic carbocations (next to a double bond) also benefit from resonance.
- Hyperconjugation: Adjacent alkyl groups can donate electron density to the carbocation through sigma bonds (hyperconjugation). More alkyl groups mean more hyperconjugative stabilization.
- Inductive Effects: Electron-donating alkyl groups stabilize carbocations. Electron-withdrawing groups destabilize them.
Experimental Observations
- Hydride Shifts: These shifts have been observed in various reactions involving hydrogen halides (HBr, HCl, HI) and alcohols.
- Alkyl Shifts: Alkyl shifts occur in reactions where rearrangement leads to more stable carbocations.
- Product Analysis: Analyzing reaction products provides evidence for rearrangements.
Ozonolysis
- Ozonolysis is an organic reaction where unsaturated bonds (such as carbon-carbon double bonds) are cleaved using ozone (O₃).
- During ozonolysis, multiple carbon-carbon bonds are replaced by carbonyl (C=O) groups, resulting in the formation of aldehydes, ketones, or carboxylic acids.
- While it is predominantly applied to alkenes (double bonds), alkynes and azo compounds can also undergo cleavage via ozonolysis.
Mechanism of Ozonolysis
Imagine an alkene (a molecule with a carbon-carbon double bond). When ozone reacts with this alkene, it breaks the double bond, leading to the formation of an intermediate called an ozonide. The ozonide can then be further processed through different workup steps to yield various products:
- Reductive Workup: In this case, the ozonide is treated with a reducing agent (such as dimethyl sulfide or zinc/acetic acid). Resulting products include aldehydes and ketones.
- Oxidative Workup: Here, the ozonide is treated with hydrogen peroxide (H₂O₂) or other oxidizing agents. The products obtained are carboxylic acids.
Electrophilic Addition Reactions of Alkenes
Alkenes, with their carbon-carbon double bonds (π-bonds), are highly reactive due to the presence of π electrons. Electrophilic addition reactions occur when an electrophile (an electron-deficient species) attacks the π-bond, leading to the addition of new atoms or groups across the double bond. These reactions are fundamental in organic chemistry and play a crucial role in synthesizing various organic compounds. Here are the key aspects of electrophilic addition reactions:
General Reaction
During electrophilic addition, the π bond of the alkene is broken, resulting in the formation of two new sigma (σ) bonds. The electrophile (the attacking species) is attracted to the π electrons in the double bond. The overall outcome is the addition of atoms or functional groups to the alkene.
Mechanism
Let’s take the example of the reaction between an alkene (such as propene) and a protic acid (HX, where X is a halogen):
- Step 1: Electrophilic Attack: The electrophile (H⁺ from the acid) attacks the π electrons of the alkene. This forms a carbocation intermediate (a positively charged carbon).
- Step 2: Nucleophilic Attack: A nucleophile (often a halide ion, X⁻) donates its lone pair to the carbocation. The X⁻ ion bonds to the positively charged carbon, completing the addition.
Overall, the π bond breaks, and two new σ bonds (C-X and C-H) form.
Representing Organic Reactions
- Organic chemists often represent reactions without balancing equations.
- Quantitative yields are less critical; the focus is on understanding the reaction pathway.
- For example, if a series of reactions leads to a desired product, chemists consider the overall efficiency rather than individual yields.
Application to Alkynes
- Alkynes (triple bonds) also undergo electrophilic addition reactions.
- If the reagent is in a one-to-one mole ratio, it adds to one of the π-bonds.
- Excess reagent can lead to a second addition, forming a doubly substituted product.
Markovnikov’s Orientation
- Markovnikov’s Rule (also known as Markownikoff’s Rule):
- Formulated by Russian chemist Vladimir Markovnikov in 1870.
- Describes the outcome of some addition reactions to unsymmetrical alkenes.
- Statement: In the addition of a protic acid (HY) to an unsymmetrical alkene, the hydrogen (H) tends to add to the carbon atom with fewer hydrogen substituents (i.e., the less substituted carbon).
- The resulting product is called the Markovnikov product.
- Example: When HBr adds to 1-butene, the major product is 2-bromobutane (following Markovnikov’s rule).
Free Radical Addition Reactions of Alkenes
Protons (electrophiles) are not the only species that initiate addition reactions to carbon-carbon double bonds (alkenes). Conflicting reports on the regioselectivity of HBr additions led to the discovery of alternative radical chain reactions.
Anti-Markovnikov Product Formation
Peroxide-Initiated Reaction
- Peroxides (such as H₂O₂) break homolytically, forming alkoxy radicals.
- The alkoxy radical abstracts a hydrogen atom from HBr.
- A bromine atom is formed and adds to the π-bond of the alkene, giving the more stable carbon radical.
- Another hydrogen abstraction generates the anti-Markovnikov alkyl bromide and a new bromine atom.
This free radical chain addition competes favorably with slower ionic additions (especially in non-polar solvents). Note that HBr is unique in this respect; the radical addition process is unfavorable for HCl and HI due to endothermic steps.
Polymerization
- The best-known use of free radical addition to alkenes is polymerization.
- Concentrated solutions of alkenes readily undergo radical-initiated polymerization.
- For example, propene can polymerize via radical addition to form polypropylene.
Anti-Markovnikov Orientation
- The anti-Markovnikov rule describes regiochemistry where a substituent is bonded to the less substituted carbon in an addition reaction.
- This is contrary to Markovnikov’s rule.
- The process is unusual because carbocations (commonly formed during alkene reactions) tend to favor the more substituted carbon.
Markovnikov’s rule guides the addition of protic acids to alkenes, while free radical addition reactions can lead to anti-Markovnikov products. These concepts play a crucial role in understanding reaction outcomes and designing synthetic routes.
Summary
We began by unraveling elimination reactions, distinguishing between E1 (unimolecular) and E2 (bimolecular) mechanisms. E1 reactions involve carbocation formation, while E2 reactions are concerted processes. Factors like base strength, solvent choice, substrate stability, and stereochemistry influence their outcomes. Zaitsev’s rule guided us toward the more substituted alkene product. Next, we ventured into the world of ozonolysis, where ozone (O₃) cleaves double bonds. This process transforms alkenes into aldehydes, ketones, or carboxylic acids. Fun fact: ozonolysis has a quirky backstory—it’s also known as “Harries ozonolysis.” Our journey continued with electrophilic addition reactions of alkenes. Electrophiles eagerly attacked π-bonds, leading to the formation of two new σ bonds. Markovnikov’s rule dictated that hydrogen tends to add to the less substituted carbon, yielding the Markovnikov product. But wait! Free radical addition reactions defied convention—anti-Markovnikov products emerged, especially when peroxides were involved.
For more regular updates you can visit our social media accounts,
Instagram: Follow us
Facebook: Follow us
WhatsApp: Join us
Telegram: Join us
1 thought on “Elimination Reactions- E1 and E2 reactions”